Mês de conscientização da Fibrose Cística: onde estamos em relação ao tratamento?

Apesar de no Brasil o mês da Fibrose Cística ser oficialmente o Setembro Roxo, nos Estados Unidos da América (EUA), maio é o mês oficial de conscientização da Fibrose Cística. Ocorrem durante o mês diversos eventos para angariar fundos, honrar a memória de pacientes, celebrar os que estão vivos e o apoio da comunidade aos pacientes que vivem com Fibrose Cística. Outra peculiaridade dos EUA é que no dia 5 de junho comemora-se o “65 Roses Day” (Dia das 65 rosas), com o mesmo objetivo do mês de maio, em homenagem a Richard “Ricky” Weiss, um menino de 4 anos que ouvindo o nome da sua doença pela primeira vez, não conseguiu pronunciar “cystic fibrosis” e pronunciou “65 roses”. É uma oportunidade para relembrarmos também a batalha e o caminho que foi trilhado para que a ciência chegasse ao ponto atual de tratamento da doença e as barreiras que ainda precisam ser vencidas no manejo destes.
“Ai da criança cujo beijo na testa tem gosto salgado, pois ela está amaldiçoada e em breve morrerá”. Esse provérbio europeu (fazendo referência ao “beijo salgado” da Fibrose Cística e talvez a forma da época de inferir algum diagnóstico semelhante) do século XV reflete o que foi o destino de muitos pacientes que vivem com Fibrose Cística até algumas décadas atrás: devido às infecções respiratórias de repetição, desnutrição proteico-calórica muitos pacientes não chegavam à idade adulta. Até os anos 1990, a sobrevida média era de 25-30 anos, mesmo em países com maior acesso a recursos. Com a melhoria do entendimento microbiológico pulmonar, aprimoramento dos tratamentos para erradicação de Pseudomonas aeruginosa, prevenção de exacerbações, antibióticos inalatórios, anti-DNAse inalatório, suporte nutricional, psicológico houve melhora da sobrevida concomitantemente, mas até o início da década 2010, ainda não havia um tratamento específico aprovado para a fisiopatologia principal da Fibrose Cística (mutação do gene CFTR que leva à produção de proteína CFTR defeituosa ou mesmo não chega a produzi-la).
Em 2012 foi aprovada pela Food and Drugs Administration (FDA) a primeira droga cujo alvo era a via de produção da CFTR: o Ivacaftor. Essa medicação funciona como um potenciador da proteína CFTR na superfície da membrana plasmática e em pacientes com mutação de “gating” (a proteína chega até a superfície, porém permanece “fechada” ou inoperante), como G551D. Nesses pacientes houve redução das exacerbações, melhora da qualidade de vida, redução dos níveis de cloro no suor (comprovação biológica da sua eficácia!) e melhora da função pulmonar. Todavia, apesar de ser a terceira mutação mais comum da FC, apenas 3-5% dos pacientes possuem mutação de “gating”, limitando o sucesso do tratamento específico. São seis classes de mutação (Figura 1), sendo mutações de classe I as que não produzem nada de proteína CFTR por parada prematura de sinalização do mRNA (mutações nonsense), mutações de classe II em diante produzem a proteína CFTR, mas por alteração do DNA ocorre incorporação inadequada de aminoácido na proteína CFTR, que pode ser defeituosa dentro da membrana plasmática ou chegar à superfície plasmática e não funcionar adequadamente (mutações missense). Surgiram então as medicações corretoras da CFTR, lumacaftor e depois tezacaftor, que combinadas com o ivacaftor também tratavam os pacientes homozigotos para F508del, abrangendo um número maior de pacientes (F580del é a mutação mais comum da FC, presente uma cópia em aproximadamente 80% dos pacientes e homozigose em torno de 50% dos pacientes). A primeira medicação que se demonstrou capaz de melhorar função pulmonar, reduzir cloro no suor, reduzir exacerbações e melhorar qualidade de vida em pacientes heterozigoto para F580del foi o elexacaftor + tezacaftor (dois corretores) + ivacaftor, aprovado pela FDA em 2019 e no Brasil, incorporado pelo SUS em 2023 e disponibilizado para a população em 2024.
Desde o surgimento dos moduladores de CFTR, temos visto uma revolução no tratamento da FC: redução no número de hospitalizações, melhora na qualidade de vida, pacientes saindo da lista de transplante pulmonar e um verdadeiro suspiro para pacientes que antes tinham expectativas e perspectivas de vida diferentes. Realmente estamos vivendo uma nova era de tratamento da FC para muitos pacientes, conseguindo reduzir exposição a antibióticos, medicações inalatórias em alguns pacientes, trazendo outra realidade para as pessoas que vivem com Fibrose Cística. No entanto, ainda muitos pacientes com mutações classe I não são candidatos a nenhuma terapia com moduladores de CFTR e a pergunta da comunidade científica é: quais os próximos passos?
As terapias que foram desenvolvidas e/ou estão em desenvolvimento incluem “read through agents”, medicações que permitiram o ribossomo “ignorar” o stop codon prematuro e permitir a transcrição da proteína. O Ataluren, medicação utilizada na Distrofia de Duchenne que auxilia na preservação de função muscular, foi tentado para FC com resultados inconsistentes e os estudos fase III foram negativos. Outras moléculas com objetivo semelhante estão em estudo, ainda sem resultados concretos. Terapias gênicas, terapias com mRNA, terapias de “gene editing” e plasmídio inalatório estão em estudo, algumas dessas técnicas se demonstrando promissoras, mas ainda é muito cedo para apontar qual delas será o caminho adequado no futuro do tratamento da Fibrose Cística. O que sabemos atualmente é que o cenário é animador e que teremos novidades interessantes para os pacientes portadores de Fibrose Cística nos próximos anos e quanto mais médicos, tanto da atenção primária/básica quanto da especializada, estiverem conscientes em relação à importância da suspeita diagnóstica, da investigação da doença e da existência de terapias novas atuais para a doença, melhor será o cuidado com esses pacientes.
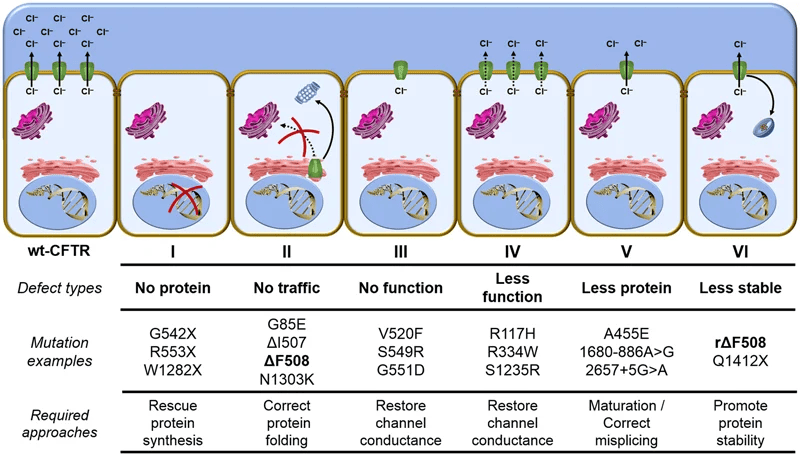
Figura 1. Esquematização das seis classes de mutações da proteína CFTR na Fibrose Cística¹.
Referências Bibliográficas
- Lopes-Pacheco M. CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front Pharmacol. 2016
- A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. Ramsey et al. NEJM 2011.
- Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. Jain et al. NEJM 2019.
- https://www.cff.org/intro-cf/cf-awareness-month
Homero Rodrigues dos Passos, pneumologista pelo HCFMUSP, Fellow de Doenças de Vias Aéreas.
 PneumoPapers
PneumoPapers
 PneumoPapers
PneumoPapers
 PneumoPapers
PneumoPapers PneumoPapers
PneumoPapers PneumoPapers
PneumoPapers