Além do Idiopático – Como a genética mudou os rumos das Doenças intersticiais

O Novo Paradigma das Doenças Intersticiais
A Pneumologia contemporânea atravessa uma transição histórica fundamental: a metamorfose das Doenças Pulmonares Intersticiais (DPI) de condições classificadas pelo rótulo da “idiopática” para entidades molecularmente mapeadas. A incorporação da genética e da biotecnologia à prática clínica já não pertence apenas ao imaginário da ficção científica — como a transformação de Anakin Skywalker em Darth Vader no Episódio III de Star Wars, marcada pela fusão entre biologia e tecnologia — mas representa hoje uma necessidade estratégica para superar a antiga passividade terapêutica. Afinal, “tempo é pulmão”.Nesse cenário, a Fibrose Pulmonar Idiopática (FPI), definida rigorosamente pelos critérios das diretrizes ATS/ERS/JRS/ALAT, permanece como o arquétipo das DPIs progressivas e irreversíveis, porém, o cenário de doença ‘’ Idiopática’’ vem perdendo espaço a cada dia.
Nossa compreensão evoluiu do foco na cicatriz fibrótica para a falência da unidade regenerativa pulmonar. A transição para uma medicina de precisão exige que olhamos além da histologia, integrando biomarcadores como o gene MUC5B e a biologia dos telômeros para oferecer o que chamamos de “novo fôlego” ao prognóstico — uma abordagem onde a intervenção é ditada pela arquitetura genômica e celular de cada paciente.
A Arquitetura Genética: O Papel Central do Polimorfismo MUC5B
Imagine que o gene MUC5B é uma “fábrica” que produz muco para proteger os pulmões, e o “promotor” é o botão de volume que controla o ritmo dessa fábrica.
O polimorfismo (conhecido na genética como rs35705950) é apenas uma pequena variação comum no DNA desse botão — uma troca de uma única pecinha (a letra G pela letra T). Esse pequeno “erro de fábrica” cria um grande problema: ele trava o botão no “volume máximo”.
O polimorfismo no promotor do gene MUC5B (rs35705950) consolidou-se como o principal fator de risco genético para a FPI. Contudo, a interpretação clínica deste marcador exige uma análise sofisticada das evidências mais recente, que desafia o mundo acadêmico.
Análise Técnica e a Evolução do “Paradoxo MUC5B”:
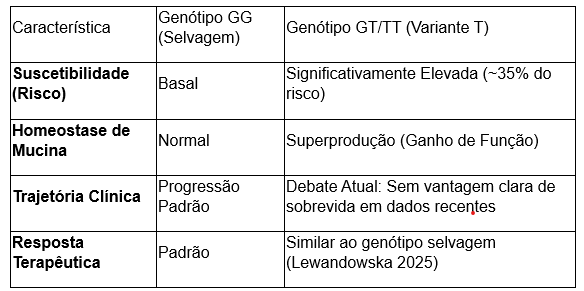
- Frequência Alélica (MAF): Embora estimativas genéricas apontem para 10% na população global, estudos de coorte rigorosos, como os de Lewandowska (2025) e Mouawad (2025), identificaram uma Frequência do Alelo Menor (T) de até 38,2% em pacientes com FPI, reforçando sua robustez como marcador de suscetibilidade em populações de ascendência europeia e do Oriente Médio.
- Mecanismo Patofisiológico: O polimorfismo resulta em um ganho de função, promovendo a superprodução de mucina 5B nos bronquíolos terminais e espaços aéreos distais. Esse excesso compromete o clearance mucociliar, gerando um estresse crônico no epitélio alveolar que desencadeia a cascata fibrótica.
- O Debate Acadêmico sobre Sobrevida: Historicamente, descreveu-se uma “sobrevida paradoxal” onde portadores do alelo T apresentariam um curso de doença mais lento. Todavia, dados recentes de Lewandowska (2025) demonstram que, em certas coortes, o genótipo não influenciou a trajetória da doença, a sobrevida ou o efeito do tratamento antifibrótico. Essa evidência sugere que o impacto do MUC5B pode ser mais matizado e dependente de interações genéticas secundárias.
Tabela 1: Dinâmica Genotípica na FPI (Perspectiva 2025)
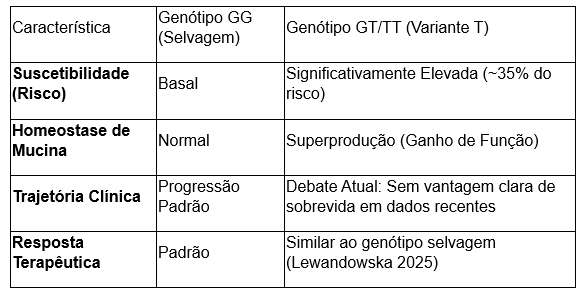
A falha regenerativa sugerida pelo risco do MUC5B converge para um pilar ainda mais crítico da patogênese: a integridade dos telômeros.
Telomeropatias e a Falência Progenitora das Células Alveolares Tipo II (AT2)
Sabe aquela pontinha de plástico que fica no final do cadarço do seu tênis ? Ela tem uma função muito importante, impedir que seu cadarço desfie e se desgaste. O telômero é a ‘’ pontinha de plástico’’ do nosso DNA, impedindo o material genético de sofrer danos durante a divisão celular.
Nas “síndromes de telômeros curtos” — causadas por mutações em genes de manutenção como TERT e TERC — a incapacidade de divisão das células progenitoras AT2, reconhecida hoje como a unidade central da regeneração pulmonar, dispara a senescência celular e a apoptose, ou seja, as células ‘’envelhecem ‘’ rápido por uma falha no mecanismo de reparação dos telômeros.
A ciência translacional avançou significativamente com o modelo do “Duplo Insulto” (Two-Hit Hypothesis). Conforme demonstrado por Povedano (2018), telômeros curtos isoladamente podem não desencadear a fibrose; é a sinergia entre a deficiência regenerativa genética e um insulto externo (como o dano por bleomicina ou estresse ambiental) que colapsa a arquitetura pulmonar.
A inovação terapêutica surge com o uso de vetores AAV9-Tert. Em modelos pré-clínicos, o alongamento dos telômeros via terapia gênica não apenas reduziu marcadores de dano ao DNA (como γH2AX), mas promoveu uma reprogramação profunda do transcriptoma celular. Dados de Povedano indicam a regulação negativa das vias TGF-β e Wnt, além da inibição das cascatas de receptores de FGF (especificamente FGFR2C e FGFR4) após o tratamento com Tert, resultando na reversão parcial da fibrose e restauração da capacidade proliferativa das células AT2.
Evolução Terapêutica: Dos Antifibrótico Convencionais ao Nerandomilast
O manejo farmacológico atual baseia-se na estabilização do declínio funcional. Os antifibróticos de primeira linha, Nintedanibe e Pirfenidona, elevaram a sobrevida média de 3-5 anos para 5-7 anos. A Pirfenidona atua primariamente na modulação do TGF-β, enquanto o Nintedanibe inibe múltiplos receptores de tirosina quinase.
O Nerandomilast emerge como a nova fronteira. Como um inibidor da fosfodiesterase 4 (PDE4), seu papel é voltado para a modulação de vias inflamatórias e fibróticas específicas, visando a estabilização funcional com um perfil de tolerabilidade distinto.
Quadro: Perspectivas de Tratamento e Mecanismos Moleculares
Medicina de Precisão e o Impacto dos Gatilhos Ambientais (Vírus)
A integração do modelo “Two-Hit” é essencial para entender por que apenas alguns indivíduos com predisposição genética manifestam a doença. A meta-análise de Sheng (2019) confirmou que infecções virais persistentes por EBV, CMV, HHV7 e HHV8 aumentam drasticamente o risco de iniciação da FPI. Crucialmente, essa associação ocorre de maneira independente de idade ou gênero, atuando como o gatilho que dispara a falência epitelial em pulmões com telômeros encurtados.
Para o futuro, a medicina de precisão foca na correção direta de erros genéticos. Tecnologias de Base Editing e Prime Editing oferecem a possibilidade de corrigir mutações específicas sem causar quebras de fita dupla no DNA. Um exemplo crítico é a abordagem das mutações no gene ABCA3, essencial para a produção de surfactante nas células AT2. Devemos diferenciar clinicamente as mutações de Classe 1 (erros de endereçamento intracelular/mistrafficking) das de Classe 2 (prejuízo na hidrólise de ATP), pois cada uma pode exigir estratégias distintas de edição gênica para restaurar a capacidade progenitora das células alveolares.
O Futuro das Doenças Intersticiais
Estamos abandonando o niilismo terapêutico para entrar na era da restauração funcional. A convergência entre o mapeamento de variantes como o MUC5B, a compreensão da falência progenitora por Telomeropatias e a mitigação de gatilhos virais redefine nossa capacidade de intervenção.
3 Pilares para a Próxima Década na Pneumologia Intersticial:
- Genotipagem Sistemática: Incorporação do status MUC5B e mensuração de telômeros para estratificação de risco e personalização de protocolos.
- Terapia de Manutenção Progenitora: Evolução dos antifibróticos atuais para terapias de ativação telomerásica e inibição das vias Wnt/FGF.
- Vigilância do “Segundo Insulto”: Monitoramento rigoroso e controle de infecções virais crônicas como estratégia de prevenção primária em indivíduos de risco.
O “Novo Fôlego” para a Pneumologia não reside na mera supressão da inflamação, mas na nossa capacidade científica de reativar o potencial regenerativo do epitélio pulmonar e proteger a linhagem celular AT2 contra o colapso molecular.
Braulio Nunes, Pneumologista pelo HCFMUSP, Fellow de Doenças Intersticiais Pulmonares
 PneumoPapers
PneumoPapers
 PneumoPapers
PneumoPapers
 PneumoPapers
PneumoPapers PneumoPapers
PneumoPapers